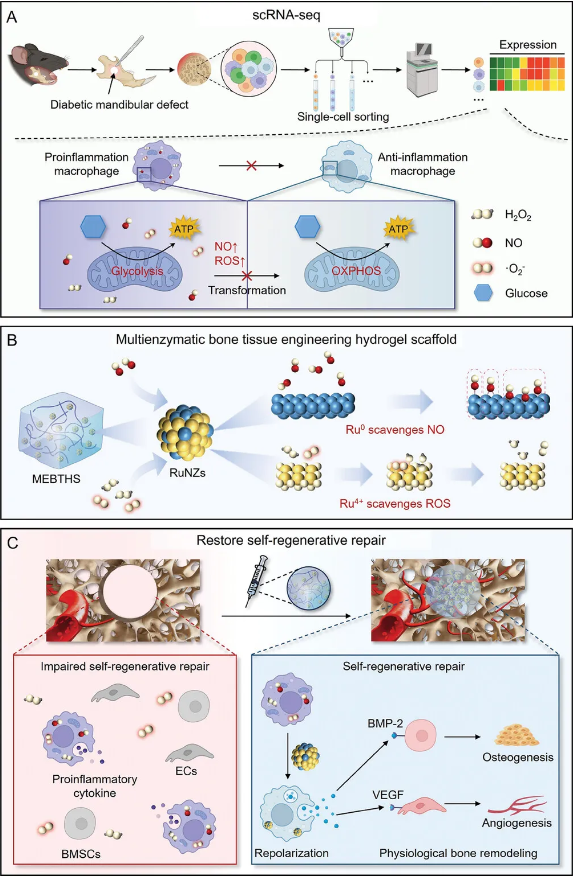
AM:上海交通大学李方园/凌代舜/浙江大学俞梦飞合作制备多酶水凝胶促进糖尿病下颌骨缺损的自我再生修复
由于促炎巨噬细胞向抗炎巨噬细胞的复极化受损,传统的骨组织工程材料难以在糖尿病期间恢复生理性骨重塑。2024年10月22日,上海交通大学李方园、凌代舜和浙江大学俞梦飞共同通讯在Advanced Materials 在线发表题为“A Single-Cell RNA Sequencing Guided Multienzymatic Hydrogel Design for Self-Regenerative Repair in Diabetic Mandibular Defects”的研究论文。该研究利用单细胞RNA测序(scRNA-seq)技术,揭示了糖尿病生理性骨重塑过程中,一氧化氮(NO)和活性氧(ROS)在阻碍巨噬细胞复极化过程中的关键作用。基于此,作者设计了一种多酶骨组织工程水凝胶支架(MEBTHS),该支架由甲基丙烯化明胶水凝胶与钌纳米酶整合而成,具有Ru0和Ru4+成分,有助于通过Ru0高效消除NO,同时Ru4+表现出ROS清除性能。因此,MEBTHS通过中和ROS、逆转NO介导的线粒体代谢来协调巨噬细胞重编程,从而使糖尿病下颌缺损内的骨髓间充质干细胞和内皮细胞恢复活力,产生质量与正常骨相当的新生骨。该研究制备的多酶水凝胶促进了缺损骨的自我再生修复,标志着骨组织工程的重大进步。外伤、颌面肿瘤切除、放化疗或牙周炎感染引起的下颌骨缺损严重影响生活质量,合并糖尿病(DM)时影响更为严重。由于炎症反应的持续和加剧发生,糖尿病的存在增加了治疗失败的风险,损害了糖尿病微环境中的生理性骨再生。因此,迫切需要开发能够促进糖尿病下颌骨缺损愈合的先进骨组织工程材料。下颌骨的生理性骨重建是一个精细调整的过程,包括巨噬细胞复极、血管生长和骨髓间充质干细胞(BMSCs)的成骨分化。然而,糖尿病期间,骨微环境发生了显著变化,促炎性巨噬细胞长期积聚且促炎表型向抗炎表型的转化受损,严重阻碍成骨和血管生成。虽然现有的骨组织工程材料在清除活性氧(ROS)方面成效显著,恢复了骨生成和血管生成中包含的BMSCs和内皮细胞(ECs)活性,但在调控巨噬细胞表型转变方面仍存在一定劣势。因此,深入了解糖尿病下颌骨缺损中促炎巨噬细胞向抗炎巨噬细胞复极的机制对于设计骨组织工程材料至关重要。图1 MEBTHS协调巨噬细胞重编程以促进糖尿病下颌骨缺损的自我再生修复示意图(摘自Advanced Materials )一氧化氮(NO)作为一种多效性细胞内信使,在炎症调节中起着至关重要的作用。研究表明,NO可以抑制碳流向氧化磷酸化(OXPHOS),炎巨噬细胞依赖线粒体代谢过程,而促炎巨噬细胞依赖于糖酵解。因此,NO通过影响细胞内线粒体代谢来阻碍巨噬细胞从促炎表型向抗炎表型的转变。然而,一些研究提出了相互矛盾的结果:NO是巨噬细胞分泌炎症介质的负调节因子,促进促炎巨噬细胞向抗炎巨噬细胞的复极化。受NO在疾病微环境中浓度和位置影响,NO的不同功能对设计促进糖尿病性下颌骨缺损愈合的骨组织工程材料构成了相当大的阻碍。因此,有必要阐明NO在糖尿病下颌骨缺损巨噬细胞中的特殊功能。单细胞RNA测序(scRNA-seq)技术具有高通量、单细胞分辨率的特点,允许在单细胞水平分析转录组表达水平,有望充分揭示巨噬细胞的异质性和变化,以及糖尿病微环境中的细胞间相互作用。该研究利用scRNA-seq技术,揭示了糖尿病微环境中骨重塑期间巨噬细胞产生过量NO,抑制了线粒体代谢从糖酵解到OXPHOS的转变,从而抑制了促炎表型向抗炎表型的复极化。此外,ROS水平升高促进巨噬细胞的促炎极化,导致血管生成和成骨受损,为双重清除NO和ROS以促进糖尿病下颌骨缺损的愈合提供了直接证据。因此,作者通过整合生物相容性甲基丙烯化明胶(GelMA)水凝胶(Gel)和多酶钌纳米酶(RuNZs),制备了一种多酶骨组织工程水凝胶支架(MEBTHS)。利用表面Ru0和Ru4+成分,RuNZ可有效消除Ru0中的NO,同时Ru4+清除ROS,使MEBTHS具有NO和ROS清除能力。值得注意的是,MEBTHS通过线粒体代谢调节促进巨噬细胞复极化,促进BMSCs的成骨分化和ECs的血管生成分化。此外,MEBTHS保护BMSC和ECs免受氧化应激。最终,恢复生理性骨骼重塑过程,使合并糖尿病患者的下颌骨能够自我再生修复,形成与正常骨骼非常相似的新生骨。https://onlinelibrary.wiley.com/doi/full/10.1002/adma.202410962?saml_referrer
声明:本网站所有内容来源注明为“新医事”,版权均属于新医学事所有,未经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“新医事”。本网注明来源为其他媒体的内容为转载,或系自媒体发布的内容,仅系出于传递更多信息之目的,转载仅作观点分享,版权归原作者所有,如有侵犯版权,请及时联系我们。